1.1 What is the difference between the two versions (Cat. No. 015 and 016) of the QuantSeq kit?
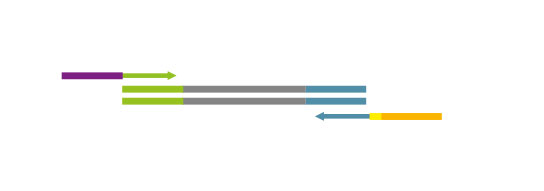
Both kit versions yield sequences close to the 3’ end of transcripts. The difference is in the location of the Read 1 linker sequence. If it is located in the 5´ part of the second strand synthesis primer (QuantSeq Forward (FWD), Cat. No. 015), NGS reads will be generated towards the poly(A) tail (Figure 1). This version is recommended for gene expression analysis.
 Figure 1: Read orientation for QuantSeq FWD (Cat. No. 015).
Figure 1: Read orientation for QuantSeq FWD (Cat. No. 015).
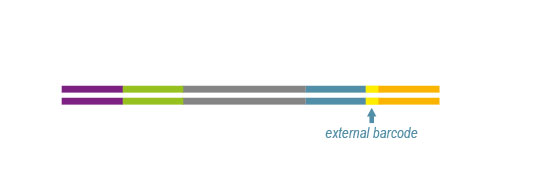
With QuantSeq Reverse (REV, Cat. No. 016) the Read 1 linker sequence is located on the 5’ end of the oligodT primer and a Custom Sequencing Primer (CSP, included in the kit) is required for sequencing in order to start the read directly at the 3´ end (Figure 2). Based on this the exact 3’ UTR can be pinpointed.
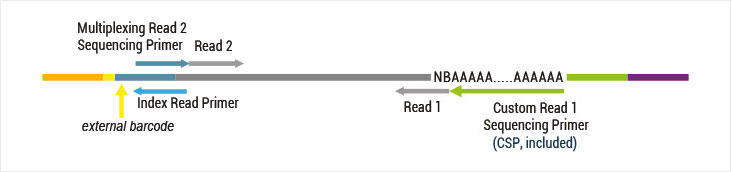
Figure 1: Read orientation for QuantSeq FWD (Cat. No. 015).
1.2 What is the CSP and how do I use it?
For QuantSeq REV (Cat. No. 016) the Read 1 linker sequence is located at the 5’ end of the oligodT primer. Here a Custom Sequencing Primer (CSP, included in the kit) is required to achieve cluster calling on Illumina machines. It covers the poly(T) stretch and replaces the Multiplex Read 1 Sequencing primer.
PROVIDE THE RELEVANT INFORMATION TO YOUR SEQUENCING FACILITY ALONG WITH THE CSP!
HiSeq 2000, HiSeq 2500 (CSP Version 2 added on cBot)
CSP Version 2 should be provided in a tube strip at 0.5 µM final concentration in a volume of 120 µl (final concentration 0.5 µM, to be diluted in HT1 = Hybridization buffer). Take 0.6 µl of 100 µM CSP Version 2 and add 119.4 µl of HT1 buffer per sequencing lane. Place the 8-tube strip into the cBot position labeled primers.
HiSeq 2500 (CSP Version 2 replaces HP10 in cBot Cluster Generation Reagent Plate)
Alternatively, CSP Version 2 can be placed directly into the cBot Cluster Generation Reagent Plate. ATTENTION: The standard Illumina Multiplex Read 1 Sequencing Primer solution HP10 (for V4 chemistry located in row 2) provided in the cBot Cluster Generation Reagent Plate has to be REMOVED first! The Illumina V4 chemistry cBot Cluster Generation Reagent Plate only has 8 rows filled. A simple trick is to have the empty rows facing towards you, this way if you want to use a CSP in lane 1, you have to remove the HP10 solution from well 1 (first one on the far left) of the 2nd row, rinse the well a couple of times with HT1 and then add the diluted CSP Version 2. For this take 1.25 µl of 100 µM CSP Version 2 and add 248.75 µl of HT1 buffer per sequencing lane. The CSP should be at 0.5 µM final concentration in a volume of 250 µl (final concentration 0.5 µM, to be diluted in HT1 = Hybridization buffer). ATTENTION: Do not add the CSP to the Standard Illumina Multiplex Read 1 Sequencing Primer = HP10 solution! Always use fresh HT1 and add the CSP / HT1 dilution to the empty and rinsed well.
HiSeq 2500 – Rapid Run
Add 12.5 µl of 100 µM CSP Version 2 to 2487.5 µl HT1 = Hybridization buffer, resulting in a total volume of 2.5 ml and a final CSP concentration of 0.5 µM. In a rapid run, both lanes will use the same sequencing primer. It is not possible to run the two lanes with different sequencing primers.
MiSeq
Clustering is performed on the machine, not on the c-Bot. The MiSeq uses a reservoir of 600 µl with 0.5 µM sequencing primer final concentration, i.e., 3 µl of 100 µM CSP Version 2 in 597 µl HT1.
HiSeq 3000, HiSeq 4000 (CSP Version 2 replaces HP10 in cBot Cluster Generation Reagent Plate)
Usage of a custom sequencing primer is currently not supported on HiSeq 3000 and 4000 machines. A work around as described for the HiSeq2500 (CSP Version 2 REPLACES HP10 in the cBot Cluster Generation Reagent Plate) is possible though. ATTENTION: Do not add the CSP Version 2 to the HP10 solution! A primer mixture would result in low clusters calls and the resulting reads would be contaminated by poly(T) stretches. Always use fresh HT1 and add the CSP Version 2 / HT1 dilution to the empty and rinsed well.
1.3 What is the typical library fragment size?
The QuantSeq protocol is optimized for shorter reads (SR50, SR100) and yields mean library sizes of about 292 – 384 bp / 477 bp (diluted SS1), with mean insert sizes of 157 – 262 bp / 355 bp (diluted SS1).
1.4 What are the input RNA requirements?
The kit uses total RNA as input, hence no prior poly(A) enrichment or rRNA depletion is required. The amount of total RNA needed for QuantSeq depends on the poly(A) RNA content of the sample in question. This protocol was tested extensively with various cell cultures, mouse tissues, and human reference RNA. Typical inputs of 500 ng total RNA generate high quality libraries. For mRNA-rich tissues (such as kidney, liver, and brain) input material may be decreased to 50 ng without adjusting the protocol. However, for most efficient detection of low abundant transcripts RNA inputs from 500 ng to 200 ng are recommended.
| Input RNA (UHR) |
SS1 in step 8 |
PS used in step 17 |
Library* |
Insert |
Library Yield |
PCR cycles |
| Start [bp] |
End [bp] |
Mean size* |
Mean size |
≥ 50 nt |
≥ 100 nt |
≥ 200 nt |
ng/μl |
nM |
| 2000 ng |
std SS1 |
72 μl |
122 |
1500 |
384 |
262 |
99% |
81% |
28% |
2.3 |
11.7 |
11 |
| 500 ng |
std SS1 |
72 μl |
122 |
1500 |
384 |
259 |
96% |
81% |
33% |
2.4 |
11.2 |
12 |
| 500 ng |
Diluted SS1 |
72 μl |
122 |
2000 |
477 |
355 |
99% |
91% |
63% |
2.0 |
7.9 |
15 |
| 50 ng |
std SS1 |
72 μl |
122 |
1500 |
349 |
227 |
98% |
78% |
27% |
3.1 |
17.0 |
15 |
| 10 ng |
std SS1 |
48 μl |
122 |
1000 |
350 |
228 |
95% |
77% |
32% |
2.1 |
10.7 |
18 |
| 5 ng |
std SS1 |
48 μl |
122 |
1000 |
292 |
170 |
94% |
66% |
17% |
2.1 |
12.1 |
20 |
| 500 pg |
std SS1 |
48 μl |
122 |
1000 |
297 |
157 |
91% |
61% |
13% |
2.8 |
17.1 |
23 |
*All libraries are prepared with external barcodes. Linker sequences are 122 bp including the 6 nt long external barcodes.
Lower inputs (10 ng or less) may require protocol adjustments, such as reducing the addition of PS in step 17 to 48 µl. Even so, an additional purification of the lane mix with 1 x PB (e.g., 50 µl lane mix plus 50 µl PB) and following the protocol from step 29 on again may be necessary, especially for less than 500 pg total RNA input to prevent sequencing through poly(A) stretches and remove all library fragments below 150 bp (inserts smaller than 38 bp). For more information regarding the input RNA requirements please consult Appendix B (p. 21).
1.5 What is the minimum input amount? Are there any recommendations for low RNA input?
QuantSeq FWD was successfully tested with as little as 10 pg of Universal Human Reference (UHR) RNA input. When using less than 1 ng of total RNA input please follow these recommendations.
- 1. Skip step 2, immediately proceed to step 3.
- 3. Extend the time of the RT in step 4 to 1 h.
- 4. Reduce the time in step 6 to 5 min at 95 °C
- 5. Use 48 µl PS in step 17.
- 6. Use 27 µl PB in step 30.
- 7. Perform qPCR to determine the exact number of cycles for the endpoint PCR
1.6 How many PCR cycles are needed to amplify the libraries?
The number of cycles for your endpoint PCR depends on the type of the RNA (tissue, organism), the RIN number and the RNA input amount. The reference values given in Appendix B, p.21 are based on Universal Human Reference RNA input and the mRNA content of other RNA sources might differ. To be on the safe side and prevent under- or overcycling of your sample we recommend performing a qPCR first. Therefore we offer a PCR Add-on Kit for Illumina (020.96) with 96 additional PCR reactions. In step 25 add another 17 µl of EB or H2O to your eluted libraries so 17 µl can be used for the qPCR and 17 µl for the subsequent endpoint PCR. Follow steps 26-27, add 5 µl of Barcode 00 (BC00) and add SYBR Green I (diluted in DMSO) to a final concentration of 0.1x and conduct at least 30 cycles to make sure the amplification reaches the plateau. Afterwards take the fluorescence value where the plateau is reached and calculate where the fluorescence is at 33% of the maximum (see Fig. 3). The value where the fluorescence reaches the maximum (plateau) is taken (13744) and the fluorescence at 33% of this values (4535) shows which cycle number is optimal for the endpoint PCRs. For the sample in Fig. 3 this would be 11 cycles, but also 12 cycles can be used if a higher yield is preferred.
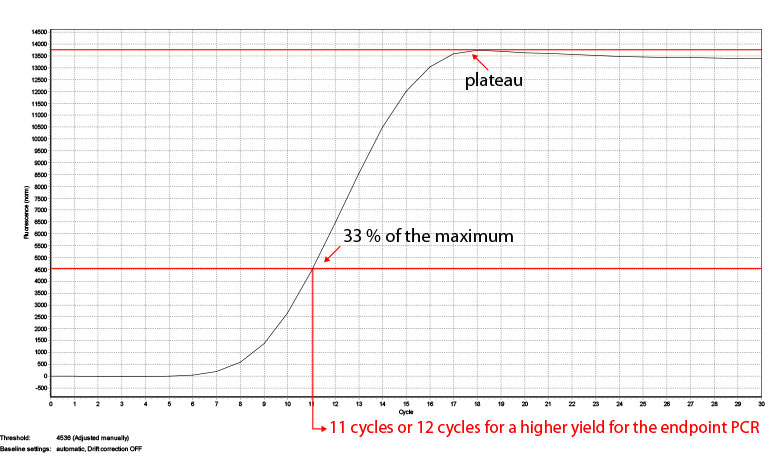 Figure 3: Calculation of the number of cycles for the endpoint PCR
Figure 3: Calculation of the number of cycles for the endpoint PCR
This cycle number is to be used for the endpoint PCR with the remaining half of your sample. Once the number of cycles for the endpoint PCR is established for one type of sample, you can use it in the following experiments and reduce the number of cycle by one as cDNA dilution for an additional qPCR is no longer required.
In the table below you can see some recommended cycle numbers for the endpoint PCR using 500 ng total RNA input for different RNA sources.
| Input RNA (500 ng) |
Cycles |
ng/µl |
nM |
| UHRR |
12 |
2.4 |
11.2 |
| HBRR |
13 |
4.0 |
20.8 |
| Hs. ES |
10 |
1.2 |
6.6 |
| M.m. heart |
13 |
3.8 |
20.9 |
| M.m. brain |
13 |
2.9 |
15.6 |
| M.m. liver |
12 |
1.3 |
6.7 |
| M.m. kidney |
12 |
2.3 |
12.2 |
| M.m. spleen |
13 |
1.4 |
8.0 |
| M.m. lung |
14 |
2.6 |
15.5 |
| M.m. ES |
11 |
1.3 |
7.5 |
| M.m. myoblast |
12 |
0.9 |
5.2 |
| M.m. fibroblast |
14 |
1.0 |
5.6 |
| M.m. myoblast progenitors |
11 |
2.1 |
11.5 |
| M.m. neural progenitors |
12 |
1.2 |
7.0 |
| Arapidopsis th. |
13 |
3.4 |
19.6 |
| Tomato seeds |
16 |
1.7 |
9.4 |
| Fungi RNA |
13 |
1.24 |
7.08 |
| Yeast RNA (S.c.) |
12 |
1.2 |
7.7 |
1.7 Is the kit suitable for preparation of libraries from degraded RNA or FFPE samples?
Yes, low quality and FFPE samples can be used with QuantSeq. Some minor protocol modification are required though:
- 1. Skip step 2, immediately proceed to step 3.
- 2.Use 48 µl PS in step 17,
- 3.use 27 µl in step 30.
Please be aware that more PCR cycles are necessary (see Table).
| ng FFPE RNA Input |
Recommended
cycle number |
| 50 ng FFPE |
18 |
| 10 ng FFPE |
24 |
| 500 pg FFPE |
27 |
1.8 How long does it take to generate QuantSeq libraries?
Lexogen’s QuantSeq kit is a library preparation protocol designed to generate up to 96 sequence-ready Illumina-compatible libraries from polyadenylated RNA within 4.5 hours. When you carry out the protocol for the first time please allow for more time and read the entire
User Guide first.
1.9 What level of multiplexing can be provided with QuantSeq? What barcoding (indexing) system do you use?
QuantSeq libraries are intended for a high degree of multiplexing. Barcodes are introduced as standard external barcodes during the PCR amplification step, allowing up to 96 samples to be sequenced per lane on an Illumina flow cell. External barcodes are 6 nt long. QuantSeq FWD libraries can be easily multiplexed with samples from other library preps. However, QuantSeq REV cannot be multiplexed with other library preps as the Custom Sequencing Primer is needed for sequencing. QuantSeq barcodes differ from Illumina’s. Exception is a barcode 5 (TAATCG) which is identical to the Illumina barcode 42.
1.10 What is the orientation of QuantSeq reads?
QuantSeq FWD (Cat. No. 015) generates NGS reads towards the poly(A) tail. To pinpoint the exact 3’ end, longer read lengths may be required. Read 1 directly reflects the mRNA sequence.
With QuantSeq REV and the custom sequencing primer it is possible to exactly pinpoint the 3’ end during Read 1. The reads generated here during Read 1 reflect the cDNA sequence, so they are in a strand orientation opposite to the genomic reference.
1.11 Which aligner should be used for data analysis?
STAR aligner or TopHat2 can be used for mapping QuantSeq FWD (Cat.No. 015) data. The reads may not land in the last exon and span a junction. For QuantSeq REV (Cat.No. 016) we do not recommend using TopHat2, since there is hardly a need to search for junctions. Nearly all sequences will originate from the last exon and the 3’untranslated region (UTR). In case of no detected junction, TopHat2 may run into difficulties. Hence, Bowtie2 or BWA can be used for mapping in this case.
More information on the data analysis can be found here.
1.12 What sequence should be trimmed?
As second strand synthesis is based on random priming, there may be a higher proportion of errors at the first nucleotides of the insert due to non-specific hybridization of the random primer to the cDNA template. These mismatches can lead to a lower percentage of mappable reads when using a stringent aligner such as TopHat2 in which case it may be beneficial to trim these nucleotides. For QuantSeq FWD (Cat. No. 015) the first twelve nucleotides need to be removed from Read 1. Alternatively, a less stringent aligner (e.g., STAR Aligner) could be used with the number of allowed mismatches being set to 14. While trimming the first nucleotides can decrease the number of reads of suitable length, the absolute number of mapping reads may increase due to the improved read quality. Reads which are too short or have generally low quality scores should be removed from the set.
While single read sequencing does not require any trimming using QuantSeq REV (Cat. No. 016) paired-end sequencing may require the first 12 nucleotides of Read 2 to be trimmed. Alternatively, also here the STAR Aligner could be used with the number of allowed mismatches being set to 16 for paired-end reads.
In case of adapter contamination detection it is crucial to trim these sequences (e.g cutadapt, trim-gallore, or bbduk) in order to align the reads.
1.13 Which sequencing platforms are suitable for QuantSeq libraries?
The QuantSeq 3’ mRNA-Seq kit (FWD, REV) are appropriate for HiSeq 2000/2500, HiSeq 3000, HiSeq 4000, GAIIX, MiSeq, while on NextSeq 500 and NextSeq 550 Illumina platforms only QuantSeq FWD can be used.
1.14 What sequencing read length is best suited for QuantSeq libraries?
Single-read 50 (SR50) sequencing runs are suitable for both QuantSeq versions. Although, for QuantSeq REV it is favorable to use longer runs (SR100), since starting the read exactly at the 3`UTR will also show the polyadenylation signal, which covers up to 25 nt from the transcription end. As the polyadenylation signal is quite similar for all transcripts it is better to have longer reads >SE 50 available for mapping in order to increase the number of uniquely mapping reads.
1.15 I want to do a SR100 or PE100 run. Are there any special considerations for this?
For paired-end sequencing we generally recommend using QuantSeq REV. Using QuantSeq FWD the quality of Read 2 would be very low due to the poly(T) stretch at the beginning of Read 2. For the library preparation itself we recommend using only 24 µl of PB in step 30 of the QuantSeq protocol (Purification, QuantSeq User Guide, page 15). In this way, inserts smaller than 50 bp are more efficiently removed and the quality of the sequencing run will improve as the likelihood of reading into poly(T) stretches decreases. For step 31-39 simply follow the
User Guide.
1.16 I see poly(T) stretches in my QuantSeq REV (Cat. No. 016) data and low sequencing quality. What is the reason for this?
For QuantSeq REV it is essential to use the CSP for Read 1 sequencing. Make sure that your sequencing facility is aware of this. When handing in QuantSeq Rev libraries for sequencing please include the CSP and the
PDF on how to use the CSP. The CSP should never be mixed together with the standard Illumina Read 1 Sequencing primer. A primer mixture would result in low clusters calls and the resulting reads would be contaminated by poly(T) stretches.
1.17 I see internal priming events. How can I prevent this?
Transcripts may have different and not yet annotated 3’ ends, which might be mistaken for internal priming events of the oligodT primer, when in fact those are true 3’ ends. Artificial spike-in transcripts such as the SIRVs or the ERCC spike.in transcripts only have one defined 3’ end, this provides the only true measure to determine internal priming. If true internal priming is detected this could be a result of mis-priming during reverse transcription for instance if the temperature before or during of reverse transcription was too low. In particular, the centrifugation step in step 2 should not be carried out at 4 °C. Spin down at room temperature! As mentioned in the general section of the User Guide unless explicitly mentioned all steps should be carried out at room temperature (RT) between 20 °C and 25 °C. To prevent mis-priming during reverse transcription the reaction temperature can also be raised to 50 °C.
1.18 What are the most critical steps in the QuantSeq library generation?
- Proper mixing of the viscous solutions (such as SS1, PB, and PS) is really important. It can be facilitated when the buffers are at room temperature and if larger volumes are used for mixing (e.g., after adding 5 µl in steps 5 and 7, use a pipette set to 15 µl or 20 µl for mixing).
- Addition of the RS1 and RS2 solutions, they have to be added in an equal amount, otherwise you will get differences in the yield.
- RS2 and SS1 have to be added in sequential order. Never mix RS2 and SS1 directly with each other as this will negatively affect the library prep.
- During the magnetic bead-based purification take care to not dry the beads too long (visual cracks will appear) as this will negatively influence the elution, but also don´t carry over traces of EtOH to the next reactions.
- Perform all steps at room temperature (including centrifugation) and don´t put your samples on a cooling block or on ice.
1.19 My Bioanalyzer trace shows a second high molecular weight peak between 1000-9000 bp. Where does it come from? Is it a problem?
A second peak between 1000–9000 bp is an indication of overcycling. The library prep has been very efficient and a lot of cDNA was generated. Hence, the PCR ran out of primers and template started to denature and reanneal improperly. This results in longer, bulky molecules that migrate at a lower speed on the Bioanalyzer chip or gels. This can interfere with exact library quantification if relying solely on the Bioanalyzer results. Therefore, a qPCR assay for exact library quantification should be used additionally if such a high molecular weight peak occurs.
For future QuantSeq library preps on similar samples reduce your PCR cycle number accordingly to prevent overcycling. Overcycling may lead to a distortion in gene-expression quantification and hence should be avoided.
1.20 There is a peak around and beyond the upper marker of the Bioanalyzer trace. Where does it come from?
A carryover of Purification Beads (PB) results in a peak around and beyond the upper marker of the Bioanalyzer. Make sure not to transfer any beads after the final elution in step 38
(Purification, QuantSeq User Guide, page 15). Leave approximately 2 µl of the eluate on the beads if a complete removal of the supernatant without beads carryover is not possible.
1.21 What can I do if my libraries are undercycled?
The PCR Add-on Kit for Illumina (Cat. No. 020) includes a Reamplification primer that can be used to add some PCR cycles for your undercycled libraries. In general, however, as QuantSeq is intended for a high degree of multiplexing undercycled libraries can still be used for preparing a lane-mix. The lane-mix may need to be concentrated if many libraries of the lanemix were undercycled.
1.22 What positive control do you recommend to use?
Universal Human Reference RNA (UHRR, Agilent) is a good positive control, the most of the reference values given in the User Guide are also based on UHRR input.
1.23 I want to do paired-end sequencing, which kit version should I use?
For PE sequencing use QuantSeq REV (Cat. No. 016). We do not recommend paired-end sequencing for QuantSeq FWD (Cat. No. 015), as the quality of Read 2 would be very low due to the poly(T) stretch at the beginning of Read 2.
1.24 Do you have any protocol recommendations to get longer insert sizes?
To get longer libraries you need to modify step 8. Dilute the SS1 Mix (Second Strand Synthesis Mix 1) 1:2 with H2O (7.5 µl SS1 + 7.5 µl H2O). Diluting SS1 may require an increase in PCR cycles hence a qPCR assay is recommended.
1.25 How much of a QuantSeq library should be loaded for sequencing?
| Sequencer |
QuantSeq FWD |
QuantSeq REV |
| HiSeq 3000 / HiSeq 4000 / HiSeqXTen |
280 pM |
280 pM |
| HiSeq 2000 / HiSeq 2500 |
10 pM |
6.5 pM |
| MiSeq |
6-15 pM |
6-15 pM |
| NextSeq 500 / NextSeq 550 |
4.5 pM |
NOT RECOMMENDED |
1.26 Which barcodes should be used for multiplexing?
Various multiplexing options are available to suit your experimental design. However, care should be taken to always use sets of barcodes that give a signal in both color channels for each nucleotide position. In detail, at least one of the two bases A or C (red channel) AND one of the two bases G or T (green channel) should be present at a given nucleotide position for all Illumina sequencers except NextSeq machines which uses a different color coding system.
In general we recommend using a complete set of 8 or 12 barcodes for multiplexing (e.g., Set 1 or Set A if the 96 reaction kit is used, respectively). However, if fewer barcodes are required also subsets of each set can be chosen.
Two samples per lane: In step use 2.5 µl of BC01 and 2.5 µl BC13 for one sample and 2.5 µl BC25 and 2.5 µl BC37 for the second. Here two barcodes are applied to each sample in order to balance the red and green laser signals.
Four samples per lane: In step use 5 µl of BC01 for one sample, 5 µl BC13 for the second, 5 µl BC25 for the third, and 5 µl BC37 for the fourth. Apply only one barcode to each sample.
Eight samples per lane: In step use all the barcodes from Set 1 (BC01/BC13/BC25/BC37/ BC49/BC61/BC73, and BC85). Apply only one barcode to each sample.
Twelve samples per lane: In step use all the barcodes from Set 1 (BC01/BC13/BC25/ BC37/BC49/BC61/BC73, and BC85) plus 4 barcodes from Set 2 (BC02/BC14/BC26, and BC38) if you have the 24 reaction kit (Cat. No. 001.24). Alternatively, if using the 96 reaction kit (Cat. No. 001.96) barcodes BC01 – 12 from row A can be used. Apply only one barcode to each sample.