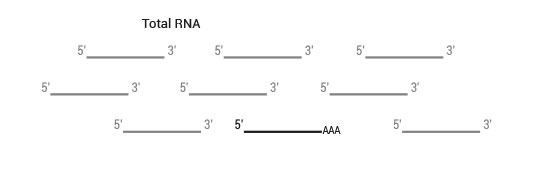
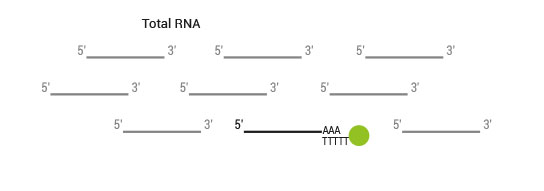
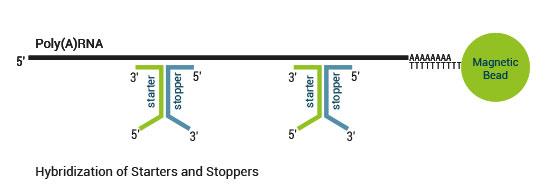
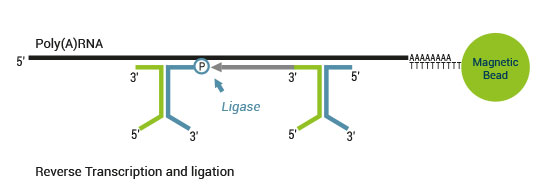
Product Description
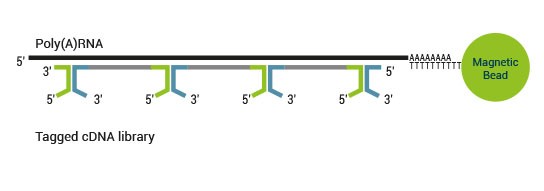
SENSE is a complete strand-specific mRNA-Seq library prep kit for accurate gene expression profiling, transcriptome sequencing, discovery and quantification of antisense transcripts and overlapping genes
The typical amount of input total RNA is 1 ng – 2 µg.
All-in-One Solution
No additional kits or reagents for poly(A) RNA selection, library amplification, size selection or purification are required or barcodes.
Different Sequencing Read Length
For good representation and even coverage of all transcripts in your experiment the library should have a size suitable for the chosen sequencing read length. The size of SENSE libraries for Illumina can be adjusted by simply modulating appropriate buffers during RT/ligation and purification steps.
Compatibility
SENSE mRNA-Seq Library Prep Kits are available for Illumina, Ion Torrent, and SOLiD sequencing platforms.
Superior Strand-Specificity
The strand-displacement stop/ligation technology used in SENSE generates fewer antisense artifacts which can be produced by template-switching in protocols which utilize RNA or cDNA fragmentation. This results in exceptional (>99.9%) strand-specificity and reduced experimental noise, enabling the detection and quantification of antisense transcripts with high confidence.
Rapid Turnaround
NGS-ready libraries can be produced from total RNA samples in under 5 hours with less than 50% hands-on time, allowing RNA extraction, library preparation and quality control to be performed in one day.
Efficient rRNA Elimination
SENSE mRNA-Seq Library Prep includes the Poly(A) RNA Selection Kit (Cat. No. 039) which virtually eliminates cytoplasmic ribosomal RNA (<0.001% of total reads from Universal Human Reference RNA) and removes the need for additional selection or depletion kits, saving you time and money.
Simple Multiplexing
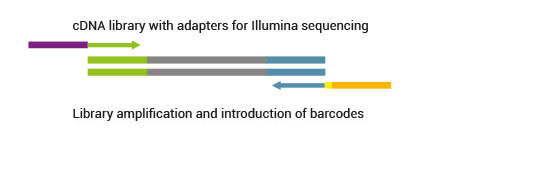
SENSE libraries can be multiplexed with very well balanced barcode sequences.Learn more about barcodes for multiplexing on different sequencing platforms.
FAQs
GeneralIllumina SpecificIon Torrent SpecificSOLID SpecificAutomation Specific
1.1 How is the exceptional strand-specificity achieved?
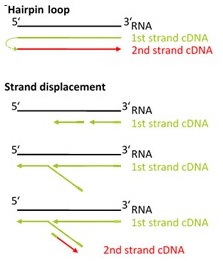
There are two major aspects of the SENSE technology contributing to its exceptional strand-specificity. Both of them are suppressing spurious second strand cDNA synthesis (see Figure 1), which introduces technical variation in detection of the antisense transcripts. Firstly, the protocol does not require RNA fragmentation as the insert size is determined by the distance between starter/stopper binding sites. Therefore, spurious second strand synthesis from the 5’ ends of RNA fragments is absent during reverse transcription. Secondly, when reverse transcriptase approaches the next hybridized primer during the first strand synthesis it is efficiently stopped, thus eliminating strand-displacement activity of the enzyme which can also cause spurious second strand synthesis.
Figure 1. Spurious second strand synthesis mechanisms. Firstly, when RT reaches 5’ ends it adds 1-5 nucleotides in a non template fashion, like a terminal transferase. When it happens the primer binding domain is free again and it can associate with a new primer, then RT flips back onto the first strand cDNA in a hairpin loop manner and a spurious second strand is initiated from this 5’ end. Secondly, when one random hexamer is extended it displaces at least to some degree the extension product of the second hexamer. This “free” displaced first strand can be further primed again with a random primer and thus create a spurious second strand.
The SENSE technology avoids both hairpin loop and strand displacement artifacts, providing the basis for the excellent strand-specificity.
1.2 Why do you use ERCC spike-in controls to determine strandedness?
ERCC spike-in controls are sets of artificial transcripts with known strand orientation which do not contain any antisense transcripts. Therefore all detected antisense ERCC reads can be considered false positives introduced during library preparation. In contrast, genome wide calculations of strandedness are conflated by true antisense transcription. Therefore true strand specificity can only be calculated on ERCC data, providing threshold levels for distinguishing endogenous antisense transcript levels from spurious second strand synthesis background.
1.3 How long does it take to generate SENSE libraries?
SENSE mRNA-Seq: 8 ready-to-sequence libraries can be prepared in under 5 hours starting from total RNA.
autoSENSE mRNA-Seq: With autoSENSE 96 libraries can be prepared on a liquid handler within 9 hours, including ~2 hours preparation time and manual interventions.
Allow for more time when you carry out the protocol for the first time and perform QC (such as Bioanalyzer).
1.4 Do I need to purchase additional kits, e.g., for ribosomal RNA depletion, size selection, library amplification, and purification?
All SENSE mRNA-Seq kits already include the Poly(A) RNA Selection Kit (Cat. No. 039), all enzymes for reverse transcription, ligation, and PCR, as well as magnetic beads for purification and the barcodes for multiplexing.
1.5 Can I still perform the protocol without thermomixer?
You can also run the protocol without thermomixer. Just mix the reverse transcription and ligation reaction from time to time with a pipette. Alternatively you can also get the Poly(A) RNA Selection Kit (Cat. No. 039) in combination with the SENSE Total RNA-Seq kit which does not require a thermomixer.
1.6 Is the kit suitable for preparation of libraries from degraded RNA or FFPE samples?
We do not recommend using FFPE samples for SENSE mRNA-Seq. The protocol is performed on oligodT beads, hence the presence of a poly(A) tail of the RNA is essential. For optimal sequencing results we recommend using RNA with RIN score of 8 or greater. Libraries generated from low quality RNA may have a 3’ bias in sequencing results. However, SENSE Total RNA-Seq can be used on low quality samples (including FFPE).
1.7 What equipment is needed for performing the SENSE protocol?
SENSE mRNA-Seq: For preparation of the SENSE library the following equipment is needed
(User-supplied Consumables and Equipment, SENSE User Guide (page 7)):
- Magnetic rack
- Benchtop centrifuge (12,000 x g, rotor compatible with 1.5 ml tubes or 96-well plates))
- Calibrated single-channel pipettes for handling 1 µl to 1000 µl volumes
- Thermomixer for 1.5 ml tubes (dry bath incubator with shaking function)
- Thermocycler
- UV spectrophotometer to quantify RNA
- Recommended: Bioanalyzer (Agilent Technologies) for library quantification. Alternative quantification methods are: qPCR assays, Nanodrop or Qubit measurements
autoSENSE mRNA-Seq: For preparation of the SENSE library the following equipment is needed (User-supplied Consumables and Equipment, autoSENSE mRNA-Seq Library Prep Kit for Illumina on PE Sciclone/Zephyr User Guide (page 12)):
- Magnetic rack (for manual bead wash)
- Microplate centrifuge
- Agencourt® 96-ring magnet (1 for Sciclone, 1 for Zephyr, PE: CLS 128316)
- Magnet spacer (1 for Sciclone, PE: CLS 133514)
- Inheco 96-well adapters (2 for Sciclone, 1 for Zephyr, PE: CLS 128372)
- Inheco 96-well adapter/shaker (1 for Sciclone, 1 for Zephyr, PE: CLS 100852)
- Thermocycler (Bio-Rad HSP-96 PCR-plate compatible)
- UV-spectrophotometer to quantify RNA
- Recommended: Automated microfluidic electrophoresis station Agilent Technologies 2100 Bioanalyzer, PerkinElmer LabChip GX II). Alternative quantification methods are: qPCR assays, Nanodrop or Qubit measurements
1.8 How complex are SENSE libraries? How is complexity being assessed?
Based on our experience the best way to assess complexity is by generating discovery plots (Figure 2).
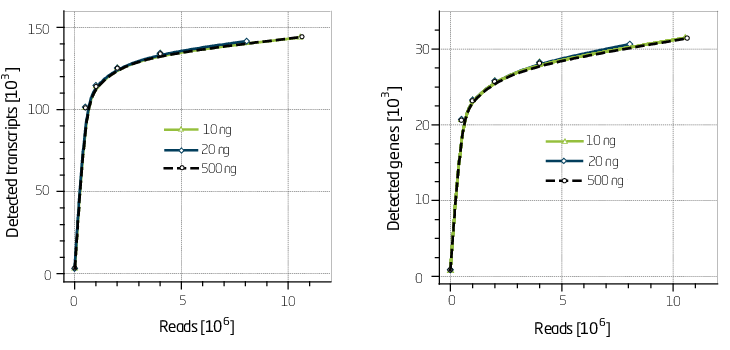
Figure 2. Discovery plots for 10 ng, 20 ng and 500 ng of input Universal Human Reference RNA (UHRR).
SENSE libraries have an excellent discovery rate on the transcript as well as the gene level.
1.9 What is the orientation of SENSE reads?
SENSE libraries are suitable for single- and paired-end sequencing. In contrast to most other library preparation protocols, SENSE libraries generate reads in a strand orientation opposite to the genomic reference. Reads must be re-oriented during data processing, either by generating the reverse complement before mapping or by inverting the directionality flag in the alignment files after mapping. Read 2 generates sequences corresponding to the original RNA molecule.
For sequencing details we refer to
Appendix F: Data Analysis, SENSE mRNA-Seq Library Prep Kit V2 User Guide (page 30).
1.10 Why do I see a second peak in high molecular weight regions (between 1,000 - 9,000 bp) on the Bioanalyzer traces?
The peak in these regions (Figure 3) is an indication of overcycling. Performing the qPCR reaction to determine the cycle number of your endpoint PCR as recommended in
Appendix A: RNA Requirements – PCR Cycles, SENSE mRNA-Seq Library Prep Kit V2 User Guide (page 19) should prevent overcycling. Still, even overcycled PCRs can be used for subsequent sequencing reactions without compromising your results. However, for further experiments using the same input RNA please adjust your cycle number accordingly or take advantage of the qPCR option.
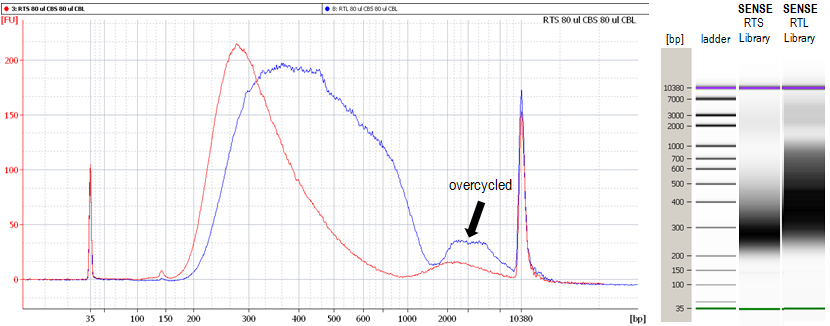
Figure 3. Bioanalyzer traces of RTS (red) and RTL (blue) synthesized SENSE for Illumina libraries with a second peak in high molecular weight regions due to overcycling.
1.11 SENSE mRNA-Seq shows very little cytoplasmic rRNA reads (0.0004%) but why do I still see mt-rRNA reads?
The low percentage of cytoplasmic ribosomal RNA demonstrates the exceptional specificity of the poly(A) selection in the SENSE protocol. Mitochondrial rRNAs, however, are polyadenylated and hence will also be selected when using the oligodT beads.
1.12 How many PCR cycles are needed to amplify the libraries?
The number of cycles for your endpoint PCR depends on the type RNA (tissue, organism), the RIN number and the RNA input amount. The reference values given in Appendix B, p.23 are based on Universal Human Reference RNA input and the mRNA content of other RNA sources might differ. To be on the safe side and prevent under- or overcycling of your sample we recommend doing a qPCR first. Therefore each SENSE Kit contains 8 additional PCR reactions. For more reactions we offer a PCR Add-on Kit for Illumina (020.96) with 96 additional PCR reactions. In step 33 add another 17 µl of EB or H2O to your eluted libraries so 17 µl can be used for the qPCR and 17 µl for the following endpoint PCR. Follow steps 35-37 using the Barcode 00 (BC00) and add SYBR Green I in a final concentration of 0.1x (diluted in DMSO) and conduct at least 30 cycles to make sure the amplification reaches the plateau. Afterwards take the fluorescence value where the plateau is reached and calculate where the fluorescence is at 33% of the maximum. This cycle number is to be used for the endpoint PCR with the remaining half of your sample. Once the number of cycles for the endpoint PCR is established for one type of sample, you can use it in the following experiments and reduce the number of cycle by one. For an example, see Fig. 4. The value where the fluorescence reaches the maximum (plateau) is taken (13744) and the fluorescence at 33% of this values (3435) shows which cycle number is optimal for the endpoint PCRs. In case of the sample in Fig. 4 this would be 11 cycles, but also 12 cycles can be used if a higher yield is preferred.
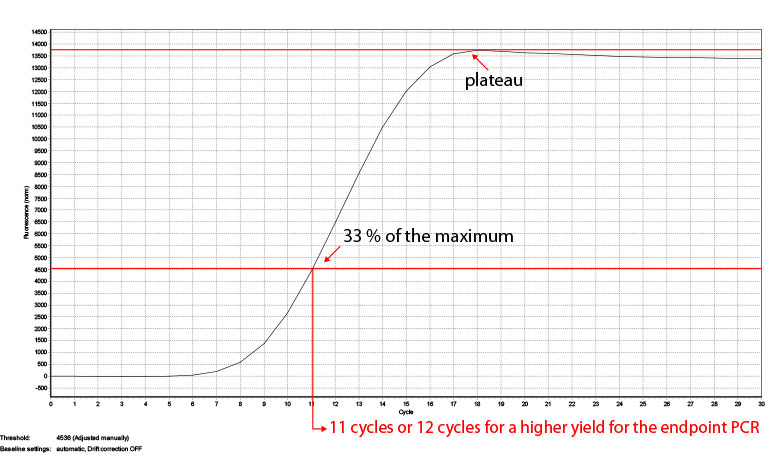
Figure 4: Calculation of the number of cycles for the endpoint PCR
1.13 What positive control or standards are you recommending to use?
Universal Human Reference RNA (UHRR, Agilent) is a good positive control, the most of the reference values given in the User Guide are also based on UHRR input.
2.1 Which sequencing platforms are suitable for SENSE libraries?
SENSE mRNA-Seq and autoSENSE mRNA-Seq Library Prep Kits V2(001.08, 001.24, 001.96) are appropriate for HiSeq1000/1500,2000/2500, 3000, 4000, GAIIX, MiSeq, and NextSeq 500, 550 Illumina platforms.
2.2 What are the input RNA requirements?
Using 1 ng – 2 µg of total RNA input from various human, animal and plant tissues, fungi and Universal Human Reference RNA yielded high quality libraries.
2.3 Are there any protocol adjustments needed for low input RNA?
For 1 – 50 ng of total RNA input the primer hybridization in step 14 should be extended to 20 minutes as well as the reaction time for the reverse transcription and ligation reaction in step 16 which should be extended to 2 hours.
2.4 What is a typical library fragment size?
SENSE mRNA-Seq Library Prep Kit V2 is offered with two different reverse transcription and ligation mixes (RTS and RTL) to be used in step 12 of the library generation to adjust the library fragment size.
SENSE mRNA-Seq: RTS produces libraries with shorter mean insert sizes (265 bp), while RTL generates libraries with longer mean insert sizes (413 – 485 bp).Typical results, SENSE mRNA-Seq Library Prep V2 User Guide (page 23).
autoSENSE mRNA-Seq:Without using the Automation Module (Cat. No. 024.96) RTS produces libraries with shorter mean insert sizes (208 bp), while RTL generates libraries with longer mean insert sizes (422 -545bp).Typical results, autoSENSE mRNA-Seq V2 for Illumina User Guide (page 33)For longer insert sizes the Automation Module is necessary
2.5 Can the insert size be regulated? Is SENSE suitable for longer read lengths e.g. 2x250 bp?
The size of SENSE mRNA-Seq libraries can be adjusted to the desired sequencing length, yielding mean library sizes of around 250 – 500 bp. This is accomplished by modulating the insert range of the library generated during RT/ligation and by using different ratios in the magnetic bead purification. Please consult
Appendix B: Adjusting Library Size, SENSE mRNA-Seq Library Prep V2 User Guide (page 23).
2.6 How long are the sequences hybridizing to the mRNA? Should those sequences be trimmed?
A 9 nt long random sequence of the starter and 6 nt long sequence of the stopper hybridize to the mRNA template. Trimming can be done with the same work-flow for both reads in a paired-end dataset. Please ensure that the selected tool preserves the read-pair information. The first nine nucleotides need to be removed from Read 1 (starter side), while on the stopper side it is only six nucleotides (Read 2).
While trimming the first nucleotides introduced by the starter/stopper can decrease the num-ber of reads of suitable length, the absolute number of mapping reads usually increases due to the improved read quality. Reads which are too short or have generally low quality scores should be removed from the set.
2.7 What level of multiplexing can be provided with SENSE? What barcoding (indexing) system do you use?
All SENSE kits already include up to 96 external barcodes. Barcodes are introduced as standard external barcodes during the PCR amplification step and require a separate sequencing reaction. SENSE libraries can be easily multiplexed with samples from other library preps. SENSE barcodes differ from Illumina’s. Exception is barcode 5 (TAATCG) which is identical to the Illumina barcode 42. For more information about barcodes and instructions about how to use them please consult
Appendix D: Multiplexing, SENSE mRNA-Seq Library Prep V2 User Guide (page 26).
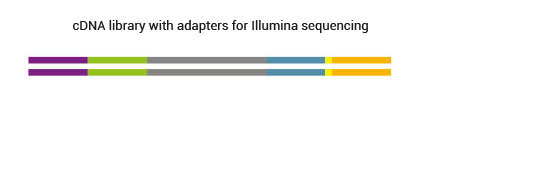
2.8 Are adapters of SENSE libraries platform-specific? Which primers should be used for sequencing?
Final SENSE mRNA-Seq libraries contain platform-specific adapter sequences. For the sequencing the standard sequencing primers can be used.
2.9 What can I do if my libraries are undercycled?
The PCR Add-on Kit for Illumina (Cat. No. 020) includes a Reamplification Primer that can be used to add some PCR cycles on top of your undercycled libraries.
3.1 What is a typical library fragment size?
SENSE is offered with two different reverse transcription and ligation mixes to be used in step 12 of the library generation. RTS will produce libraries with shorter mean insert sizes (132-189 bp), resulting in libraries ranging from 116 bp to 700 bp, while RTL generates libraries with longer mean insert sizes (269-382 bp), resulting in library fragments from 115-1700 bp.
Typical results, SENSE for Ion Torrent, User Guide (page 23).
3.2 Can the insert size be regulated?
The size of SENSE libraries can be adjusted to the desired sequencing length. This is accomplished by modulating the insert range of the library generated during RT/ligation and by using different size cut offs during purification. Please consult
Appendix B: Adjusting Library Size, SENSE User Guide (page 21).
3.3 How long are the sequences hybridizing to the mRNA? Should those sequences be trimmed?
A 6 nt long random sequence of the starter and 6 nt long sequence of the stopper hybridize to the mRNA template. As SENSE is based on random priming, there may be a higher proportion of errors at the first nucleotides of the insert due to non-specific hybridization of the starters and stoppers to the RNA. These mismatches can lead to a lower percentage of mappable reads when using a stringent aligner, in which case it may be beneficial to trim these nucleotides. The first six nucleotides of the insert should be removed from Read 1 (starter side). In case of a non-multiplexed library this means the first 10 nts (TCAG plus 6 nts of the starter) of the read should be trimmed. If the insert size was smaller than the sequencing length it might be beneficial to also trim the last 6 nts before reading into the P1 adapter sequence.
3.4 Are the adapters of SENSE libraries the same as Ion Torrent’s? Which primers should be used for sequencing?
Final SENSE libraries contain Ion Torrent adapter sequences and can be used with standard sequencing reagents.
3.5 What level of multiplexing can be provided with SENSE? What barcoding (indexing) system do you use?
SENSE libraries can be multiplexed with in-line barcodes introduced at the beginning of each read during the RT/ligation step with the SENSE In-line Barcode Kit (Cat. No. 007.08A/B or 007.24A/B), allowing up to 24 samples to be sequenced on a single Ion chip. Indexing is performed by replacing the starter/stopper mix used during reverse transcription and ligation with starter/stopper mixes supplied with the barcode kit.
4.1 What is a typical library fragment size?
SENSE for SOLiD libraries have an average library size of 244 bp with a size range of 125 – 700 bp. Of this length, adapter sequences (including a 10 nt external barcode) consume 93 bp. Total insert size therefore ranges from approximately 32 bp to 607 bp for standard SENSE libraries, with 64 % of fragments having an insert of 100 bp or greater.
Typical results, SENSE for SOLiD User Guide (page 22).
4.2 Can the insert size be regulated?
No.
4.3 How long are the sequences hybridizing to the mRNA? Should those sequences be trimmed?
A 6 nt long random sequence of the starter and 6 nt long sequence of the stopper hybridize to the mRNA template. From our experience trimming of the reads is not necessary when using LifeScope. However, as SENSE is based on random priming, there may be a higher proportion of errors at the first nucleotides of the insert due to non-specific hybridization of the starter/stopper heterodimer to the RNA. These mismatches can lead to a lower percentage of mappable reads hence it may be beneficial to trim these nucleotides. Trimming can be done with the same work-flow for both reads in a paired end dataset. Six nucleotides need to be removed from both reads (Read 1 and 2). With LifeScope it is possible to move the seed to position 6. For example to map 50 bp reads use: first.map.scheme.unmapped.50=25.2.6:15. For more information please consult the LifeScope Advanced User Guide (p.148ff).While trimming the first nucleotides introduced by the starter/stopper can decrease the number of reads of suitable length, the absolute number of mapping reads usually increases due to the improved read quality. Reads which are too short or have generally low quality scores should be removed from the set.
4.4 Are the adapters of SENSE libraries the same as SOLiD's?
Final SENSE libraries contain SOLiD adapter sequences.
4.5 What level of multiplexing can be provided with SENSE? What barcoding (indexing) system is offered?
SENSE SOLiD libraries are offered with an external barcoding option. External barcodes can be introduced during library amplification with the SENSE External Barcode Kits (Cat. No. 005.08 or 005.24, 12 barcodes, sets A-H), allowing up to 96 samples to be sequenced per SOLiD flow chip lane. External barcodes are 10 nt long. For more information about barcodes and instructions about how to use them please consult
Appendix D: Multiplexing, SENSE for SOLiD User Guide (page 23).
5.1 Which modules are needed to run the automated SENSE mRNA-Seq V2 solution on a liquid handler?
You can do automated SENSE mRNA-Seq V2 already with SENSE mRNA-Seq Library Prep Library Prep V2 for Illumina after installation of the software package, which can be easily downloaded from the download section. If you want to have a fully walk-away protocol and select for larger library sizes the Automation Module for SENSE mRNA-Seq V2 is needed in addition.
5.2 What is the advantage if using oil versus film for sealing?
If oil is used for sealing the reaction wells the Phase 1-Pre PCR of the protocol does not require any operator intervention. Alternatively, a manual film sealing can be used.
5.3 On which liquid handler is the autoSENSE protocol automated so far?
The autoSENSE mRNA-Seq protocol is currently automated on PerkinElmer Sciclone NGS workstation. The Zephyr machine is used for the post-PCR purification, which could also be done manually or on any other liquid handler on which the PostPCR SPRI Purification application is established. Lexogen is planning to automate the SENSE protocols on all other liquid handling platforms used for NGS. Please contact us if you are interested in putting SENSE on your platform.
5.4 Is it possible to use less than 96 rxn/run? Are the remaining reagents stable for later use?
The robot protocol can be set up to run for any number of columns from 1 to 12 (simply by changing the respective entry in the Excel Workbook), but it will always take the barcodes from the first N wells of the P6 plate, starting by column 1. Therefore, you can run an assay with less than 96 (for example 24) reactions in multiples of 8, but you have to prepare a new P6 plate which will substitute the barcode plate contained in the kit. You can do this by simple transferring the reagents from the kit barcode plate to the columns 1, 2, and 3 (24 reactions = 3 columns x 8 rows) of a new HSP96 plate.
The procedure is also described in detail in the
autoSENSE mRNA-Seq Library Prep Kit V2 for Illumina on PE Sciclone/Zephyr User Guide (page 22).
5.5 Should I select Yes or No in Beads P7 prewashed box?
By selecting option YES, you can save about 15 minutes of each machine run time if you do the bead washing manually in a larger batch (takes about 10 minutes for several machine runs). Typically, manual prewashing only makes sense for high throughput sample processing.
5.6 What is the purpose of the non-sterile HSP plate at B4 on the Sciclone deck?
The plate serves as a spacer to elevate the plates put on the B4 location. Because of presence of other consumables at the neighboring locations of B4, the tips on the robot head would otherwise not be able to plunge down to the bottom of the wells of a single HSP plate placed at B4.
5.7 We only have a Zephyr NGS workstation in our lab. Can we still use the protocol?
Unfortunately no. The Phase1-PrePCR of the autoSENSE protocol is currently only available for the Sciclone NGS workstation.
5.8 We have a ScicloneNGSx workstation (with sunken deck). Can we use the protocol?
The protocol is intended and programmed to run on this variant of the Sciclone liquid handler but this configuration has not been tested. Therefore, we recommend contacting Lexogen for on-site support with protocol installation.