1. How is the exceptional strand-specificity achieved?
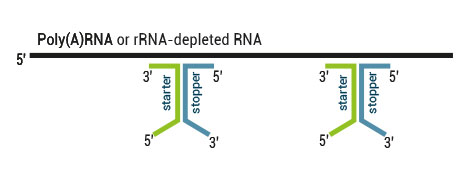
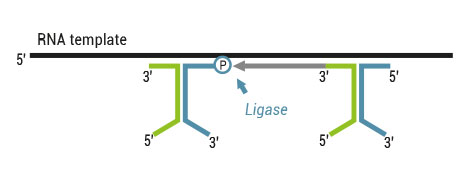
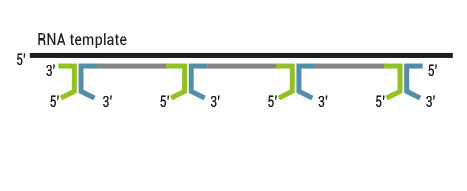
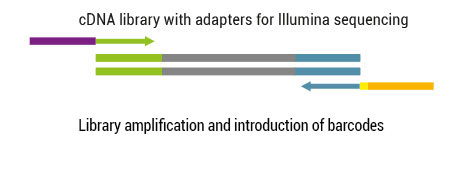
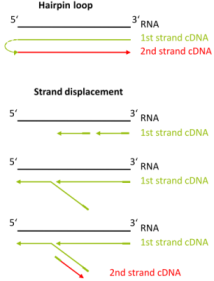
There are two major aspects of the SENSE technology contributing to its exceptional strand-specificity. Both of them are suppressing spurious second strand cDNA synthesis (see Figure), which introduces technical variation in detection of the antisense transcripts. Firstly, the insert size is determined by the distance between starter/stopper binding sites. Therefore, spurious second strand synthesis from the 5’ ends of RNA fragments is absent during reverse transcription. Secondly, when reverse transcriptase approaches the next hybridized primer during the first strand synthesis it is efficiently stopped, thus eliminating strand-displacement activity of the enzyme which can also cause spurious second strand synthesis
Spurious second strand synthesis mechanisms. Firstly, when RT reaches 5’ ends it adds 1-5 nucleotides in a non template fashion, like a terminal transferase. When it happens the primer binding domain is freed again and this can associate with a new primer, then RT flips back onto the first strand cDNA in a hairpin loop manner and a spurious second strand synthesis is initiated from this 5’ end. Secondly, when one random hexamer is extended it displaces at least to some degree the extension product of the second hexamer. This “free” displaced first strand can be further primed again with a random primer and thus create a spurious second strand.
The SENSE technology avoids both hairpin loop and strand displacement artifacts, providing the basis for the excellent strand-specificity.
2. Why do you use ERCC spike-in controls to determine strandedness?
The protocol was extensively tested with input amounts of 1 ng to 2 µg of total RNA input using RNA extracted from mouse liver, kidney, brain, lung, spleen, thymus, and heart as well as Universal Human ERCC spike-in controls are sets of artificial transcripts with known strand orientation which do not contain any antisense transcripts. Therefore, all detected antisense ERCC reads can be considered false positives introduced during library preparation. In contrast, genome wide calculations of strand-specificity are conflated by true antisense transcription. Therefore, true strand-specificity can only be calculated on ERCC data, providing threshold levels for distinguishing endogenous antisense transcript levels from spurious second strand synthesis background.
3. How long does it take to generate SENSE libraries?
8 ready-to-sequence SENSE Total RNA-Seq libraries can be prepared within 3.5 hours starting from poly(A) or rRNA-depleted total RNA. Allow for an additional 2 h for the rRNA depletion of total RNA using RiboCop or 1 hour for the poly(A) selection with the Poly(A) RNA Selection Kit, respectively.
Allow for more time when you carry out the protocol for the first time and QC (such as Bioanalyzer) if needed.
4. Do I need to purchase additional kits, e.g., for ribosomal RNA depletion, size selection, library amplification, and purification?
The SENSE Total RNA-Seq kit already includes external barcodes, all enzymes for reverse transcription, ligation, and PCR, as well as all reagents for purification. It is also available as a bundle with the RiboCop rRNA Depletion Kit (Cat. No. 042).
Magnetic beads for poly(A) selection using the Poly(A) RNA Selection kit need to be purchased separately.
5. What are the input RNA requirements for SENSE Total RNA-Seq?
Typical inputs of poly(A) selected and rRNA-depleted RNA from various tissues, plants, bacteria of 0.5 ng to 500 ng RNA will generate high quality libraries.
6. Is the kit suitable for preparation of libraries from degraded RNA or FFPE samples?
SENSE Total RNA-Seq is suitable for FFPE and low quality RNA samples.
7. What equipment is needed for performing the SENSE protocol?
For preparation of the SENSE Total RNA-Seq libraries the following equipment is needed (
User-supplied Consumables and Equipment, SENSE Total RNA-Seq Library Prep Kit User Guide (page 8)):
- Benchtop centrifuge (12,000 x g, rotor compatible with 1.5 ml tubes)
- Magnetic rack or plate
- Calibrated single-channel pipettes for handling 1 µl to 1000 µl volumes
- Thermocycler
- UV spectrophotometer to quantify RNA
- Chromatin Immunoprecipitated (ChIP) DNA
- Recommended: Bioanalyzer (Agilent Technologies) for library quantification. Alternative quantification methods are: qPCR assays, Nanodrop or Qubit measurements
8. Which sequencing platforms are suitable for SENSE libraries?
SENSE Total RNA-Seq library prep kits (009.08,009.24, and 009.96) are appropriate only for Illumina platforms (HiSeq2000, HiSeq3000, HiSeq4000, GAIIX, MiSeq, NextSeq 500, NextSeq 550).
9. What is the orientation of SENSE reads?
The SENSE libraries are suitable for single- and paired-end sequencing. In contrast to most other library preparation protocols, SENSE libraries generate reads in a strand orientation opposite to the genomic reference. Reads must be re-oriented during data processing, either by generating the reverse complement before mapping or by inverting the directionality flag in the alignment files after mapping. Read 2 generates sequences corresponding to the original RNA molecule. For sequencing details we refer to
Appendix F: Sequencing, SENSE Total RNA-Seq Library Prep Kit User Guide (page 29).
10. Why do I see a second peak in high molecular weight regions (between 1,000 - 9,000 bp) on the Bioanalyzer traces?
The peak in these regions (Figure below) is an indication of overcycling. Performing the qPCR reaction to determine the cycle number of your endpoint PCR as recommended in
Appendix A: RNA Requirements – PCR Cycles, SENSE Total RNA-Seq Library Prep Kit User Guide (page 18) should prevent overcycling. Still, even overcycled PCRs can be used for subsequent sequencing reactions without compromising your results. However, for further experiments using the same input RNA please adjust your cycle number accordingly or take advantage of the qPCR option.

Bioanalyzer traces of RTS (red) and RTL (blue) synthesized SENSE for Illumina libraries with a second peak in high molecular weight regions due to overcycling.
11. What is a typical library fragment size?
With the SENSE Total RNA-Seq kit you can choose between four different library sizes based on your sequencing needs. Depending on the combination of PS and BD used in step 10
(SENSE Total RNA-Seq Library Prep Kit User Guide (page 13)), mean library sizes between 370 – 510 bp will be produced, resulting in library fragments of 150 – 1500 bp.
12. Can the insert size be regulated? Is SENSE suitable for longer read lengths e.g. 2x250 bp?
The size of SENSE Total RNA-Seq libraries can be adjusted to the desired sequencing length, yielding mean library sizes between 370 – 510 bp. This is accomplished by modulating the insert range of the library generated after second strand synthesis by different size cut offs during magnetic bead purification. Please consult
Appendix B: Adjusting Library Size, SENSE Total RNA-Seq Library Prep Kit User Guide (page 22).
13. How long are the sequences hybridizing to the mRNA? Should those sequences be trimmed?
SENSE Total RNA-Seq uses a 9 nt long random sequence of the starter and a 6 nt long random sequence of the stopper hybridized to the RNA template. Therefore, the first nine nucleotides need to be removed from Read 1 and the first six nucleotides from Read 2.
14. What level of multiplexing can be provided with SENSE? What barcoding (indexing) system do you use?
For the SENSE Total RNA-Seq kit 96 external barcodes are available for multiplexing. They are introduced as standard external barcodes during the PCR amplification step. SENSE libraries can be easily multiplexed with samples from other library preps. SENSE barcodes differ from Illumina’s. Exception is a barcode 5 (TAATCG) which is identical to the Illumina barcode 42. For more information about barcodes and instructions about how to use them please consult
Appendix E: Multiplexing, SENSE Total RNA-Seq Library Prep Kit User Guide (page 27).
15. Are adapters of SENSE libraries platform-specific? Which primers should be used for sequencing?
Final SENSE Total RNA-Seq libraries contain Illumina-specific adapter sequences. For the sequencing the standard primers can be used.
16. What positive control or standards are you recommending to use?
Universal Human Reference RNA (UHRR, Agilent) is a good positive control, the most of the reference values given in the User Guide are also based on UHRR input.
17. What can I do if my libraries are undercycled?
The PCR Add-on kit for Illumina includes a Re-amplification primer that can be used to add some PCR cycles on top of your undercycled libraries.
18. Are there any recommendations for FFPE RNA in SENSE Total RNA-Seq?
FFPE RNA is often heavily fragmented, so STs hybridized in shorter distances, generating small (or too small) insert sizes.
- For FFPE RNA we recommend:
- 1.Dilute STs 1:10
- 2.Skip step 2
- 3.In step 10 use 15 µl PB + 13 µl BD + 27 µl PS
- 4.Optional: In step 14 add only 39 µl of PS